
HQSpectrum – Die Plattform für moderne NMR-Analyse
Physikbasierte NMR-Vorhersagen. Geräteunabhängig. Für jede Spektrometerfrequenz.
Von der Molekülstruktur zur verifizierten NMR-Analyse
HQSpectrum ist eine durchgängige Plattform für die Vorhersage, Simulation und Analyse von NMR-Spektren. Die Software begleitet den gesamten Workflow – von der molekularen Struktur bis zum fertigen Analysebericht.
Ob 60-MHz-Benchtop-NMR oder Hochfeldspektrometer mit über 600 MHz: HQSpectrum liefert nachvollziehbare, reproduzierbare und physikalisch fundierte Ergebnisse für Strukturverifizierung, Gemischanalyse und Qualitätskontrolle.
Dank quantenmechanischer Spin-System-Modelle können NMR-Spektren präzise berechnet, mit experimentellen Daten verglichen und effizient ausgewertet werden.
Ihre Vorteile
NMR-Analysen auf Expertenniveau für das gesamte Team
Zuverlässige Ergebnisse auch ohne Hochfeldspektrometer
Nachvollziehbare und reproduzierbare Resultate für Forschung, Qualitätskontrolle und regulatorische Anforderungen

Bild: ©CyrilMarcilhacy/AgenceOblique

WEBINAR
Kann ich Benchtop-NMR in meinem Labor einsetzen?
Juni, 2026 | 15:00 – 17:00 Uhr (CEST)
Warum die Auswertung von NMR-Spektren häufig zum Engpass wird
Die Auswertung von NMR-Daten erfordert häufig spezialisiertes Fachwissen und bindet wertvolle Ressourcen.
Die Interpretation von Spektren kann Experten über Stunden oder sogar Tage beschäftigen.
Bei Benchtop-NMR erschweren Signalüberlagerungen und geringere spektrale Auflösungen die Analyse zusätzlich.
Ergebnisse unterscheiden sich häufig zwischen Geräten, Anwendern und Laborstandorten.
Die Folge: Wertvolle Analysezeit geht verloren. Projekte verzögern sich, externe Dienstleistungen verursachen zusätzliche Kosten und die Skalierbarkeit analytischer Prozesse wird eingeschränkt.
Unsere Lösung
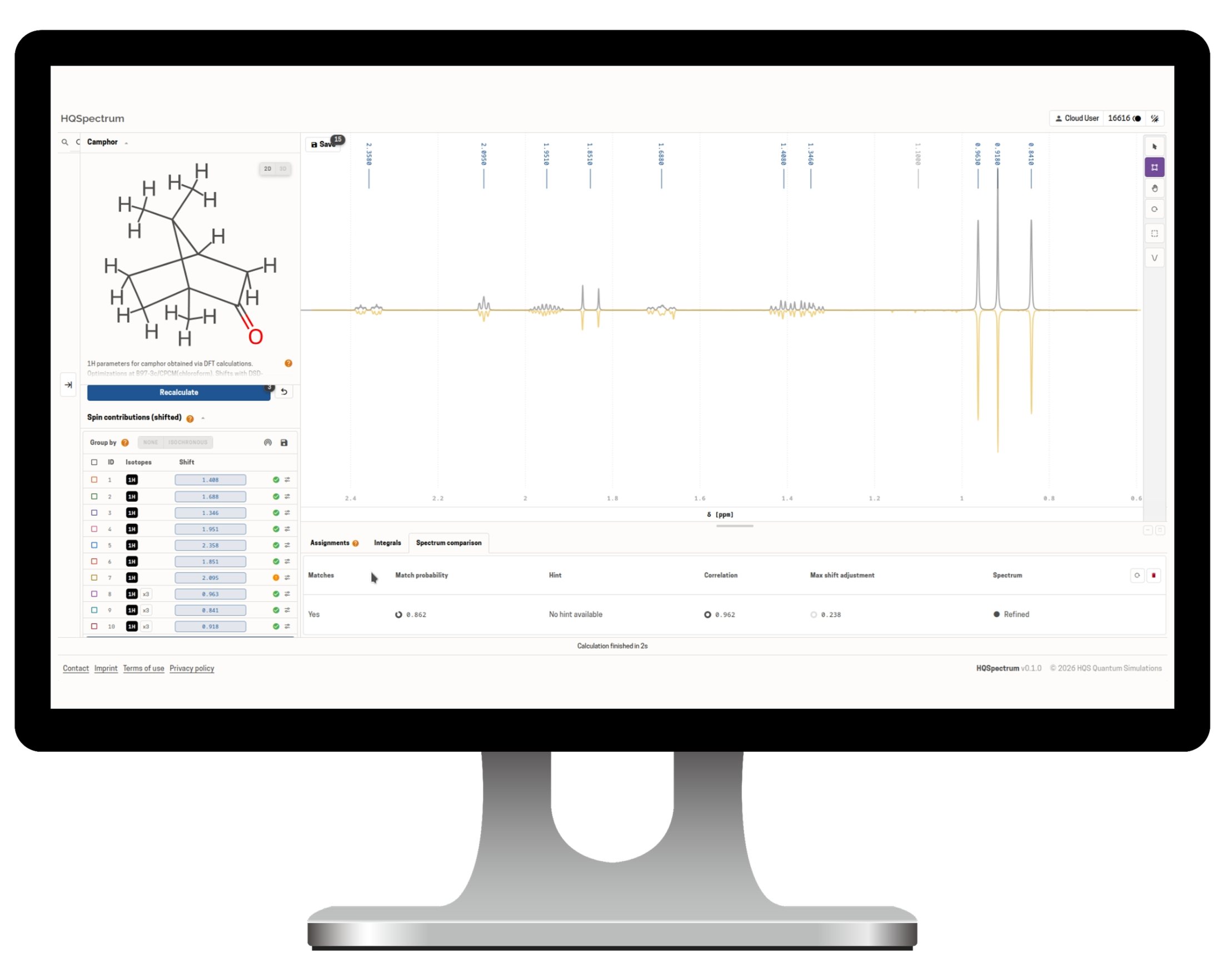
Physikbasierte NMR-Software für Benchtop- und Hochfeld-NMR
HQSpectrum schließt die Lücke zwischen kostengünstigen Benchtop-NMR-Systemen und der analytischen Leistungsfähigkeit moderner Hochfeldspektroskopie.
Im Zentrum der Plattform steht ein quantenmechanischer Solver, der chemische Verschiebungen, Kopplungskonstanten und vollständige Multiplettstrukturen über den gesamten Frequenzbereich präzise berechnet.
Dadurch lassen sich Spektren auch dort zuverlässig analysieren, wo konventionelle Methoden aufgrund von Signalüberlagerungen an ihre Grenzen stoßen.
Nachvollziehbar
Keine Black Box
Jede Vorhersage basiert auf physikalischen Modellen. Chemische Verschiebungen, J-Kopplungskonstanten und individuelle Spin-Beiträge sind vollständig nachvollziehbar und transparent dokumentiert.
Präzise
Bei jeder Frequenz
Von 60 MHz bis über 600 MHz liefert HQSpectrum präzise NMR-Spektrenvorhersagen über den gesamten Frequenzbereich. Multiplettstrukturen und chemische Verschiebungen werden auch dann korrekt berechnet, wenn herkömmliche Werkzeuge aufgrund starker Peak-Überlagerungen keine eindeutige Interpretation ermöglichen.
Automatisiert
reproduzierbare Workflows
Vom Molekül bis zum standardisierten Analysebericht. HQSpectrum automatisiert zentrale Schritte der NMR-Auswertung und sorgt für konsistente, reproduzierbare Ergebnisse.
Die Plattform eignet sich sowohl für Einzelverbindungen als auch für komplexe Gemische und reduziert die Abhängigkeit von spezialisierter NMR-Expertise.
So funktioniert HQSpectrum
Vorhersagen

Verifizieren

Anpassen

Dokumentieren
Automatisierte NMR-Workflows für Strukturverifizierung, Gemischanalyse und Qualitätskontrolle – ohne aufwendige Experteninterpretation.
Molekül eingeben oder importieren
Geben Sie eine SMILES-Struktur ein oder zeichnen Sie Ihr Molekül direkt in der Weboberfläche. HQSpectrum unterstützt den Import von 2D- und 3D-Strukturen sowie alle gängigen Molekülformate.
NMR-Parameter berechnen
Chemische Verschiebungen und J-Kopplungskonstanten werden mithilfe quantenchemischer DFT-Simulationen inklusive Konformationssampling berechnet. Da die Berechnung nicht auf vorhandene Referenzdatenbanken angewiesen ist, können auch neue und bisher unbekannte Moleküle zuverlässig analysiert werden.
Spektrum simulieren
Der physikbasierte Solver berechnet das vollständige eindimensionale NMR-Spektrum schnell und präzise für die gewünschte Messfrequenz.
Selbst bei Niederfeld-NMR werden Multiplettstrukturen korrekt dargestellt, auch in Bereichen, in denen Standardverfahren häufig versagen.
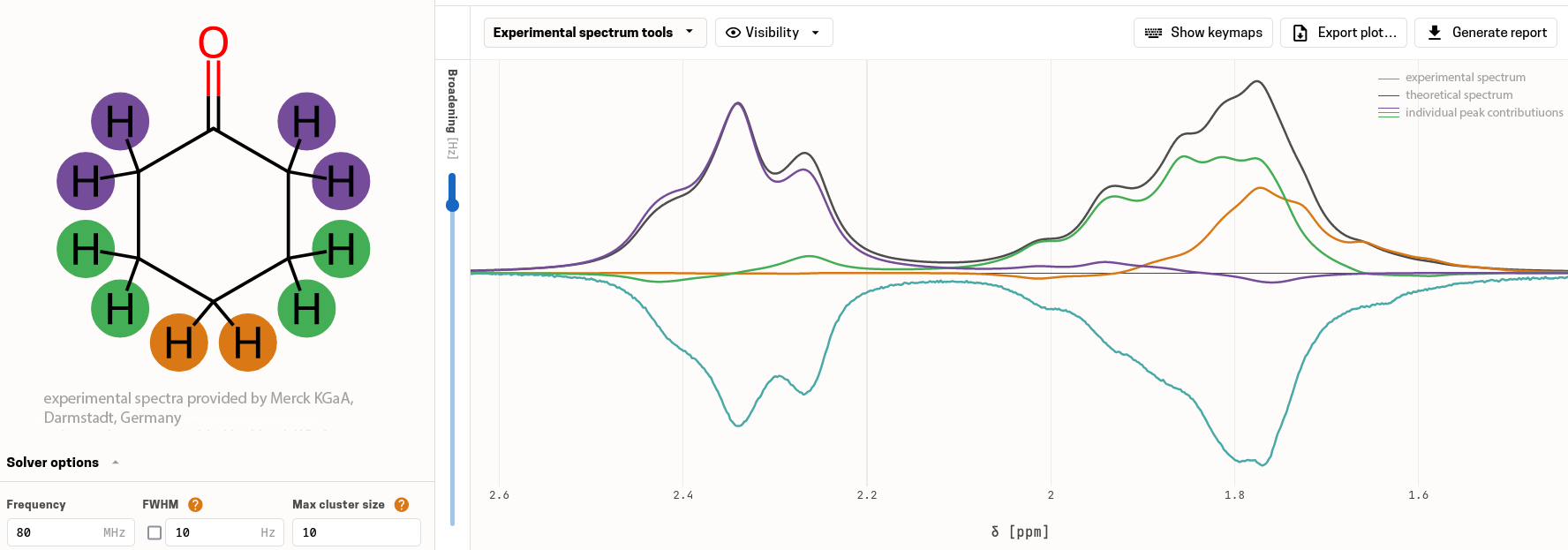
Analysieren, anpassen und verifizieren
Laden Sie Ihr experimentelles Spektrum hoch, gleichen Sie Simulation und Experiment miteinander ab, bestätigen Sie die Struktur und erstellen Sie mit wenigen Klicks einen professionellen PDF-Bericht.
Flexible Moleküleingabe
SMILES-Strings, 2D-/3D-Import oder direkte Eingabe über die Weboberfläche.Unterstützung experimenteller Spektren
Import von JCAMP-DX-, JDX- und Bruker-Dateien inklusive automatischer Bereinigung von Lösungsmittel-, Referenz- und Verunreinigungssignalen.Vollständiger Frequenzbereich
Eine Plattform für Benchtop- und Hochfeld-NMR von 60 MHz bis über 600 MHz.Peak-Zuordnung
Automatischer Abgleich von simulierten und experimentellen Spektren sowie interaktive und halbautomatische Anpassung chemischer Verschiebungen.Verifizierungsmetriken
Quantitative Bewertung anhand von Korrelationen, maximalen Verschiebungsabweichungen und weiteren Qualitätsparametern.Transparente Vorhersagen
Physikbasierte Ergebnisse auf Grundlage quantenmechanischer Modelle – unabhängig von öffentlichen Referenzdatenbanken.PDF-Berichte
Professionell aufbereitete Berichte für Dokumentation, Team-Review und Qualitätssicherung.
HQSpectrum
Funktionen im Überblick
Geplante Erweiterungen
Quantum Mechanical Spectral Analysis (QMSA)
Gemischanalyse
Strukturaufklärung
HQSpectrum wird im Rahmen des EIC-Transition-Projekts HQS NextNMR weiterentwickelt und durch den European Innovation Council (EIC) gefördert.
Im Fokus stehen insbesondere neue Funktionen zur Analyse komplexer Stoffgemische.
Lesen Sie unseren Blog und entdecken Sie weitere Anwendungsbeispiele für HQSpectrum.
Entwickelt für analytische Chemie, Spektroskopie und Forschung
Analytische Chemie
Vorhersage und Verifizierung von NMR-Spektren bekannter und unbekannter Verbindungen. Analyse überlappender Multipletts bei jeder Spektrometerfrequenz. Direkter Vergleich von Simulation und Experiment ohne manuelle Peak-Zuordnung.
Qualitätskontrolle
Automatisierte Strukturverifizierung und Verunreinigungsanalyse für routinemäßige Chargenprüfungen. Standardisierte und reproduzierbare Ergebnisse inklusive dokumentationsfähiger Berichte.
Forschung
Beschleunigen Sie Strukturaufklärung und Strukturvalidierung. Vergleichen Sie quantenmechanisch vorhergesagte Spektren mit experimentellen Daten – von einfachen organischen Molekülen bis hin zu Naturstoffen und Steroiden.
Labor- und IT-Management
Hardwareunabhängige Cloud-Plattform mit Unterstützung für JCAMP-DX-, JDX- und Bruker-Formate. Automatisierungs- und Batch-Verarbeitungsfunktionen für Hochdurchsatzlabore.
Bringen Sie Ihre NMR-Workflows auf das nächste Niveau
Testen Sie HQSpectrum kostenlos und mit vollem Funktionsumfang.
Keine Kosten. Keine Verpflichtung.
Oder sprechen Sie direkt mit unserem Team, um die passende Lösung für Ihr Labor zu finden.

Florian Wullschläger
Product Manager HQSpectrum

Dr. Konstantina Alexopoulou
Sales Lead
Wir beraten Sie gerne und zeigen Ihnen die Möglichkeiten von HQSpectrum in einer persönlichen Demonstration.
Bitte kontaktieren Sie uns per E-Mail: hqspectrum@quantumsimulations.de
Gefördert durch die Europäische Union. Die in diesem Projekt vertretenen Ansichten und Meinungen sind ausschließlich die der Autorinnen und Autoren und entsprechen nicht zwangsläufig denen der Europäischen Union oder der European Innovation Council and SMEs Executive Agency (EISMEA). Für die Inhalte übernehmen weder die Europäische Union noch die Förderbehörde Verantwortung.