
NMR Analysis Platform built on Quantum Mechanics: HQSpectrum
Physics-based NMR prediction. Hardware-agnostic. Works at all spectrometer frequencies.
NMR analysis on a new level
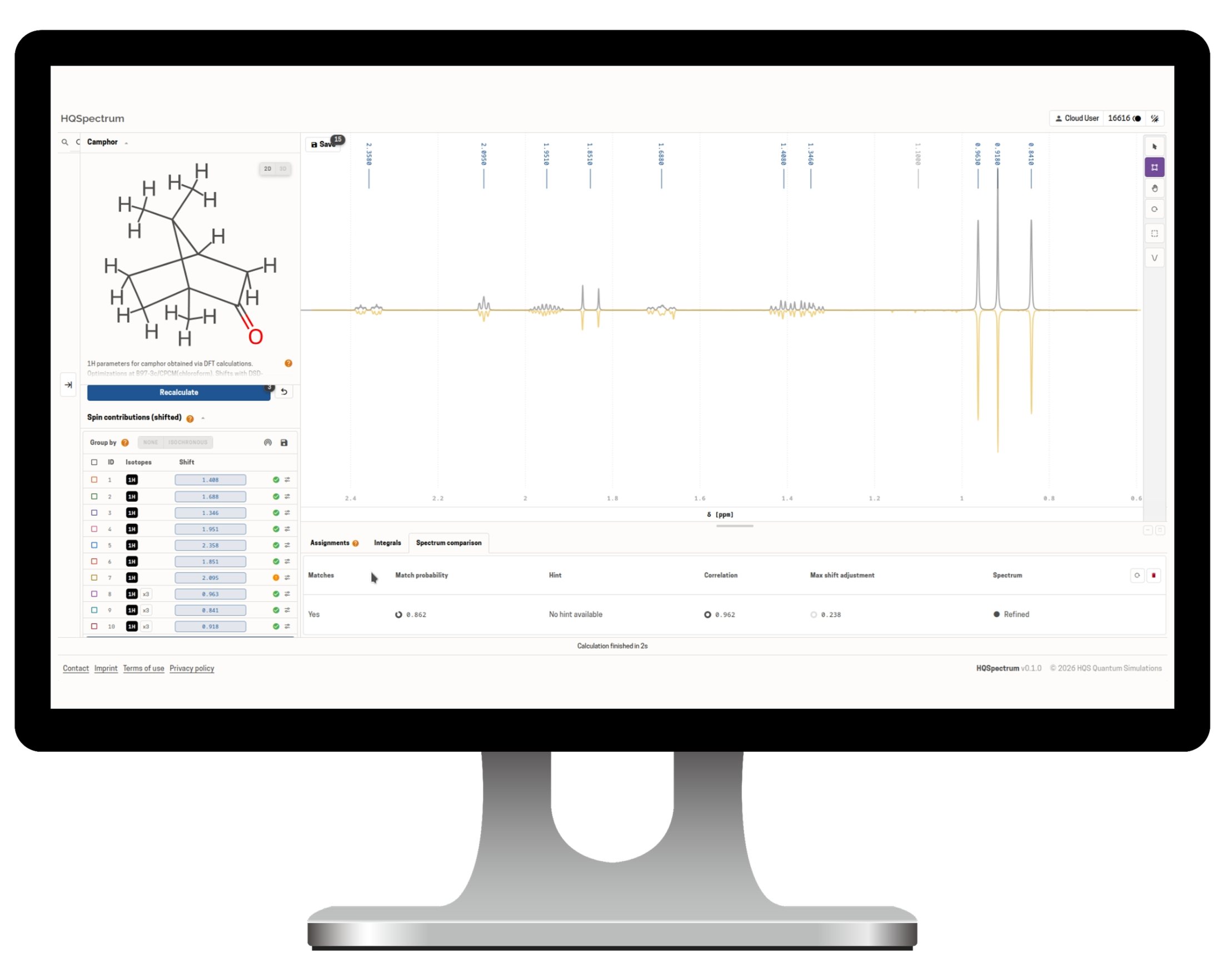
HQSpectrum is an end-to-end NMR analysis platform that takes you from molecular structure to verified report, combining one of the fastest NMR solvers available with the precision your research demands.
Predict, simulate, and interpret NMR spectra at any frequency — from 60 MHz benchtop NMR devices to 600+ MHz high-field instruments. Powered by quantum-mechanical spin-system models, HQSpectrum delivers explainable, reproducible results for structure verification, mixture analysis, and quality control workflows.
Expert-level NMR analysis for every team member.
In-house results — without high-field instrumentation.
Physics-driven outputs your team and regulators can trust.

picture: ©CyrilMarcilhacy/AgenceOblique
Why NMR Analysis Breaks Down at Scale
Spectrum interpretation ties up expert resources for hours — or days.
Benchtop NMR signals overlap where standard tools fail.
Results vary between instruments, operators, and labs.
The result: valuable analytical time and budget lost to outsourcing.
Our Solution
Physics-Based NMR Software — from Low-Field to High-Field
HQSpectrum closes the gap between affordable benchtop NMR devices and the analytical depth of high-field spectroscopy. The core is a quantum-mechanical spin Hamiltonian solver that calculates correct multiplet structures and NMR parameters at any spectrometer frequency.
Explainable
Not a Black Box
Every prediction is grounded in physics: chemical shifts, J-coupling constants, and individual spin contributions are fully traceable. Results you can justify to colleagues and regulators alike.
Accurate
at All Frequencies
Reliable NMR spectrum prediction from 60 MHz to 600+ MHz. Correct multiplet structures and chemical shift prediction where conventional tools produce unresolvable peak overlap.
Reliable
Built for Automation
From molecular structure input to standardized analytical report. Automated, reproducible, and scalable for single compounds or complex mixtures — without requiring NMR expertise.
How HQSpectrum Predicts and Analyzes NMR Spectra
Predict

Verify

Adjust

Report
Automated NMR workflows for structure verification, mixture analysis, and quality control, no expert interpretation required.
Draw or Import Your Molecule
Enter a SMILES string or draw your molecule directly in the Web UI. HQSpectrum supports 2D/3D import and all common molecular input formats.
Generate NMR Parameters
Chemical shifts and J-coupling constants calculated via ab initio DFT simulations with conformer sampling — not limited to existing public databases. Enables reliable parameter prediction for arbitrary molecules.
Simulate the Spectrum
The physics-based spin Hamiltonian solver calculates the full 1D NMR spectrum at your target frequency, fast and accurately.
Correct multiplet structures resolved even at low field, where standard tools fail.
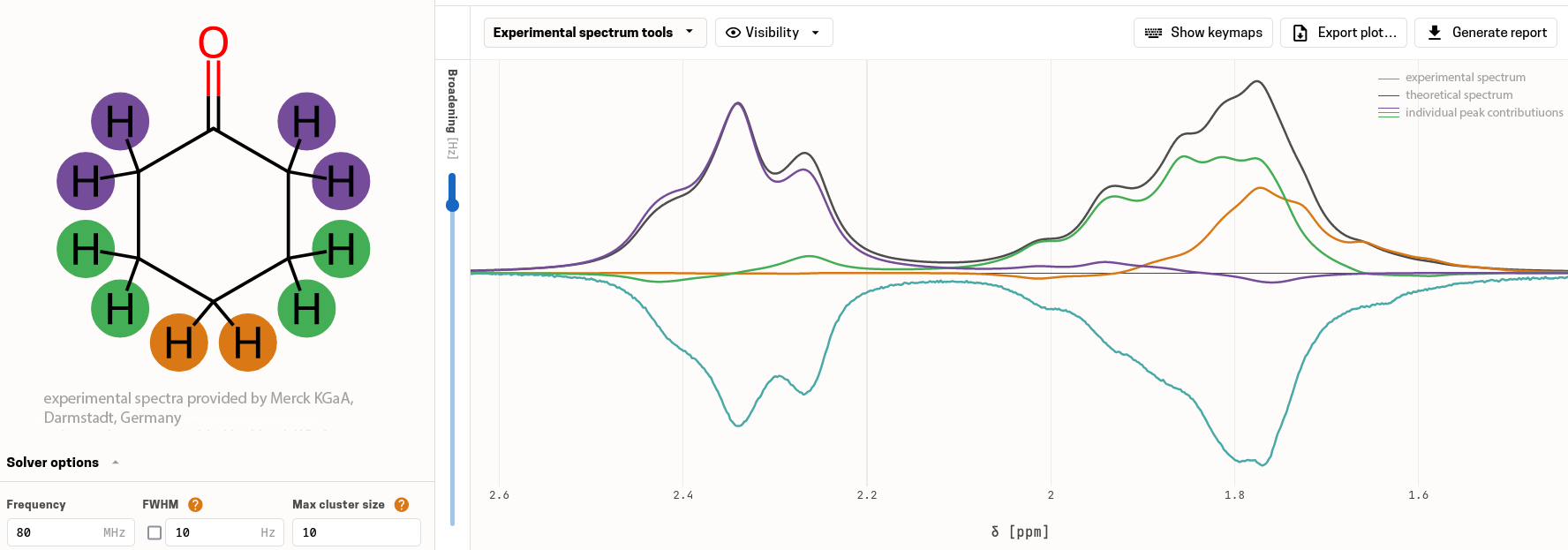
Analyze, Adjust, and Verify
Upload your experimental spectrum, align simulation and experiment, confirm your structure, and export a shareable PDF report.
Flexible Molecule Input
SMILES strings, 2D/3D import, or draw directly in the Web UI.Experimental Spectrum Support
JCAMP-DX/JDX and Bruker import. Automatic clean-up of solvent, reference, and impurity peaks.Full Frequency Range
From 60 MHz benchtop NMR to 600+ MHz high-field — one tool for every spectrometer.Nuclei-to-Peak Assignment
Automatic alignment of simulated and experimental spectra. Interactive and semi-automatic shift adjustment.Verification Metrics
Maximum shift deviation, correlation, and more. Quantitative confirmation of every structural assignment.Transparent, Explainable Predictions
Physics-based outputs grounded in quantum mechanics — not limited to existing public databases.PDF Report Creation and Sharing
Clean, shareable reports for every analysis. Ready for team review and documentation.
HQSpectrum Features at a Glance
Planned features
QMSA
mixture analysis
Structure elucidation
HQSpectrum is developed within the EIC Transition project HQS NextNMR, supported by the European Innovation Council (EIC). Within this program, HQS will expand HQSpectrums capabilities especially in the direction of analyzing mixtures.
Read our Blogarticle to explore more about use cases with HQSpectrum.
Designed for your NMR workflow: NMR Software for Analytical Chemistry, Spectroscopy, and Research
Analytical Chemists
Predict and verify NMR spectra for known and unknown compounds. Resolve overlapping multiplets at any spectrometer frequency. Compare simulated and experimental spectra side by side — no manual peak picking required.
Quality Control Labs
Automated structure verification (ASV) and impurity profiling for routine batch analysis. Standardized, reproducible outputs and shareable PDF reports for analytical workflows requiring consistent, traceable results.
Researchers
Accelerate structure elucidation and structure determination. Validate quantum-mechanically predicted NMR spectra against experimental data for complex molecules — from simple organics to natural products and steroids.
Lab & IT Managers
Hardware-agnostic cloud platform. Supports JCAMP-DX/JDX and Bruker data formats. Batch processing and automation-ready workflows for high-throughput lab environments.
Ready to Upgrade Your NMR Workflow?
Start with the free trial — full feature access,
no credit card, no commitment. Or talk to us
directly to find the right plan for your lab.

Florian Wullschläger
Product Manager HQSpectrum

Dr. Konstantina Alexopoulou
Sales Lead
We look forward to advising you and showing you our NMR analysis platform.
Please contact us at: hqspectrum@quantumsimulations.de
Funded by the European Union. Views and opinions expressed are however those of the author(s) only and do not necessarily reflect those of the European Union or European Innovation Council and SMEs Executive Agency (EISMEA). Neither the European Union nor the granting authority can be held responsible for them.